
Vínculos
Si buscas
hosting web,
dominios web,
correos empresariales o
crear páginas web gratis,
ingresa a
PaginaMX
Por otro lado, si buscas crear códigos qr online ingresa al Creador de Códigos QR más potente que existe


FARMACOCINÉTICA
La farmacocinética es la rama de la farmacología que estudia los procesos a los que unfármaco es sometido a través de su paso por el organismo. Trata de dilucidar qué sucede con un fármaco desde el momento en el que es administrado hasta su total eliminación del cuerpo.
Para ello, se han desarrollado diferentes modelos que simplifiquen los numerosos procesos que tienen lugar entre el organismo y el fármaco. Aún cuando dentro de los mismos el modelo policompartimental es el más próximo a la realidad, la complicación que conlleva ha hecho que sean los modelos monocompartimental y en todo caso el bicompartimental los más usados. Desde esos prismas, el estudio detallado de los sucesivos pasos que atraviesa el fármaco en el organismo, se agrupan bajo el anagrama LADME:
- Liberación del producto activo,
- Absorción del mismo,
- Distribución por el organismo,
- Metabolismo o inactivación, al ser reconocido por el organismo como una sustancia extraña al mismo, y
- Eliminación del fármaco o los residuos que queden del mismo.
Modelos policompartimentales
Al graficar la relación entre los distintos factores, la imagen resultante es una curva, siendo entonces necesario el cálculo de determinadas áreas bajo esa curva para hallar los resultados a las interrelaciones presentadas. Por ello, estos modelos reciben también el nombre de farmacocinética no lineal, y se basa de forma muy importante en la cinética de Michaelis-Menten. Los factores de no linealidad de una reacción, serían, entre otros, los siguientes:
- Absorción polifásica: La absorción del fármaco sigue al menos dos picos de máxima
- intensidad, con lo que mediatiza la linealidad de su llegada al plasma.
- La naturaleza del fármaco hace clara distinción entre tejidos de alta y baja irrigación.
- Saturación enzimática: En fármacos en los que su eliminación es dependiente de su biotransformación, al aumentar la dosis, las enzimas responsables de su metabolismo se saturan y la concentración plasmática del fármaco aumenta desproporcionalmente, por lo que su depuración deja de ser constante.
- Inducción o inhibición enzimática: Algunos fármacos tienen la capacidad de inhibir o estimular su propio metabolismo, en una reacción de retroalimentación. Tal es el caso de fluvoxamina,fluoxetina y fenitoína. Al administrar mayores dosis de estos medicamentos, las concentraciones plasmáticas de fármaco sin metabolizar aumenta y el tiempo medio de eliminación aumenta con el tiempo. Por esa razón, para fármacos con farmacocinética no-lineal, es necesario ajustar laposología o régimen en casos de incrementar la dosis.
- El riñón establece mecanismos activos de eliminación para algunos fármacos, independientes de los niveles de concentración plasmática.
Como se puede apreciar, la no linealidad puede venir determinada por razones que afectan a toda la secuencia farmacocinética: absorción, distribución, metabolismo y eliminación.

Modelo monocompartimental
Se le conoce como farmacocinética lineal porque al graficar la relación los distintos factores implicados (dosis, concentraciones en elplasma sanguíneo, eliminación, etcétera) la representación gráfica es una recta o una aproximación a ella. Es muy útil para fármacos que se distribuyen con rapidez desde el plasma a otros fluidos y tejidos.
El cambio de concentración respecto al tiempo viene dado por C=Cinicial*E^(-kelt) que si se representa lnC frente tiempo da lugar a una recta.
LADME
Una vez que el fármaco entra en contacto con el organismo, suceden varias fases que se reconocen con el acrónimo LADME:
- Liberación de la sustancia activa,
- Absorción de la misma por parte del organismo
- Distribución por el plasma y los diferentes tejidos,
- Metabolización, es decir inactivación de una sustancia xenobiótica y, finalmente,
- Excreción o eliminación de la sustancia o de los productos de su metabolismo.
No obstante, muchos manuales engloban la primera fase dentro de la segunda, ya que en numerosas ocasiones se administra el fármaco en forma de principio activo, con lo que ésta fase no existe. Otros hablan de una fase que engloba la distribución, metabolización y excreción que sería la «fase de disposición». Finalmente también hay autores que incluyen el aspecto toxicológico de cada fármaco en lo que se conocería como ADME-Tox o ADMET.
Cada una de las fases está sujeta a las interacciones físico-químicas entre fármaco y organismo, que se pueden expresar de forma matemática. La farmacocinética, pues, se apoya en ecuaciones matemáticas que permiten predecir el comportamiento del fármaco, y que dan cuenta, de una forma preferente, de la relación que existe entre las concentraciones plasmáticas y el tiempo transcurrido desde la administración.
La liberación es el primer paso del proceso en el que el medicamento entra en el cuerpo y libera el contenido del principio activoadministrado. El fármaco debe separarse del vehículo o del excipiente con el que ha sido fabricado, y para algunos autores comprende tres pasos: desintegración, disgregación y disolución. Se hace una especial referencia a la ionización de las moléculas del fármaco como factor limitante de la absorción, debido a las propiedades de las membranas celulares que dificultan el paso a su través de moléculas ionizadas. La recomendación de masticar los comprimidos o tabletas realizada por muchos profesionales radica, precisamente, en facilitar esta fase, en concreto la disgregación.
En todo caso, es necesario recordar que las características de los excipientes tienen un papel fundamental, ya que tienen como una de sus funciones el crear el ambiente adecuado para que el fármaco se absorba correctamente. Es por ello que medicamentos con la misma dosis, pero de distintas marcas comerciales pueden tener distinta bioequivalencia, es decir, alcanzan concentraciones plasmáticas distintas, y, por tanto, efectos terapéuticos diferentes.
Absorción
La absorción significa atravesar algún tipo de barrera, diferente según la vía de administración usada, pero que en último término se puede reducir al paso de barreras celulares. O dicho de otra forma, la interacción de la molécula con una membrana biológica, donde las características fisicoquímicas, tanto del fármaco como de la membrana, determinarán el resultado del proceso.
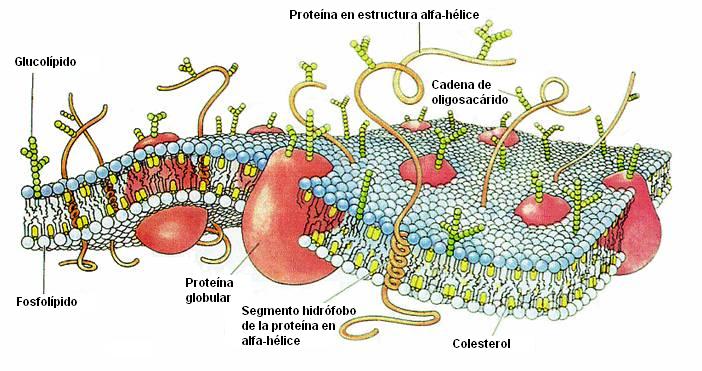
Es indispensable conocer la estructura de la membrana citoplasmática debido a su estrecha e importante relación con la farmacocinética, que implica el pasaje de las drogas a través de las membranas.
La membrana citoplasmática consiste en una capa bimolecular de lípidos, con moléculas de proteínas intercaladas, que adquiere un espesor de 75 a 80 Å (angstrom,unidad de longitud).4
Los fosfolípidos son responsables de las características depermeabilidad de la membrana así como eslabón importante en lacadena anabólica de numerosas sustancias de defensa (prostaglandinas, leucotrienos, ...). Suponen aproximadamente un 40% a 45% de los componentes de la membrana.
Por su parte, las proteínas constituyen alrededor del 50% de los constituyentes de las membranas, y le dan la rigidez estructural necesaria a la misma. Además, se comportan como el punto de inicio de las reacciones a las moléculas que llegan hasta la membrana (receptores), las metabolizan (enzimas), transportan moléculas en contra del gradiente de concentración a ambos lados de la membrana (bombas), o crean canales por donde puedan pasar éstas moléculas (proteínas canal).
Finalmente, nos podemos encontrar entre un 7% y un 10% de hidratos de carbono (glucolípidos y glucoproteínas) que actúan como modulador de las proteínas receptores.
El receptor celular es el punto último del viaje del fármaco destinado a lograr un efecto sobre el organismo humano. De las complejas interrelaciones entre ambos se encarga otra disciplina de la farmacología: la farmacodinámica.
Vías de administración
Las barreras que ha de atravesar y las características de la absorción de cada sustancia vienen determinadas por cual haya sido la vía por la que ha llegado la misma a entrar en contacto con el organismo, o dicho de otro modo, de cual sea la vía de administración. Aquí se verá sólo una breve tabla de las diferentes vías de administración, con las características especiales en cada caso de cara a la absorción.
La vía oral es la vía recomendada para humanos. Desafortunadamente, no todos los productos pueden adaptarse para su uso por esta vía. En la vía oral el fármaco llega al organismo habitualmente después de la deglución. Una vez en el estómago, se somete a las características de los jugos del mismo, que por su acidez favorece mucho la ionización del fármaco, lo que hace que la absorción sea difícil. A pesar de todo, no son escasos los fármacos que se absorben a nivel de la mucosa gástrica: los muy liposolubles, como el alcohol o ácidos débiles como los salicilatos o los barbitúricos que presentan menores niveles de ionización a pH bajo. Cuando llega el fármaco al intestino delgado cambia el pH luminal y se favorece bastante la absorción pasiva. De hecho, prácticamente todos los fármacos, menos los ácidos y basesfuertes, se absorben a este nivel. Además, en la mucosa intestinal hay numerosos mecanismos para realizar procesos de absorción en contra de gradiente, aunque difícilmente se logran niveles plasmáticos suficientes para que sean efectivos. Esta falta de absorción para algunos fármacos se aprovecha para utilizarlos a nivel local (como la neomicina o los laxantes). Igualmente, por su similitud estructural, se utiliza este efecto para administrar fármacos que no atraviesen la piel y que actúen a nivel local, constituyendo lo que se conoce como vía dérmica o vía tópica.
La vía parenteral ofrece indudables ventajas sobre la vía oral: permite su uso en pacientes que no pueden o no deben deglutir, permite el uso de sustancias polipeptídicas y otras que se inactivan por los jugos gastrointestinales y evitan el primer paso hepático. Sin embargo precisa de instrumental para su realización y presenta inconvenientes como la infección local, tromboflebitis, neuralgias, necrosis dérmicas, etc. Desde el punto de vista farmacodinámico, la principal ventaja es la facilidad para ajustar la dosis eficaz, ya que la biodisponibilidad se considera del 100% en la mayoría de los casos.
Respecto a la vía respiratoria su interés fundamental es que brinda la posibilidad de la utilización de sustancias en estado gaseoso (casi exclusivamente oxígeno o anestésicos generales). La absorción sigue las leyes del intercambio de gases a nivel alveolar y tiene la ventaja de poner en disposición una gran superficie de absorción.
Distribución
La distribución de los fármacos puede definirse, entre otras formas, como la llegada y disposición de un fármaco en los diferentes tejidos del organismo. Es un proceso muy importante, toda vez que, según su naturaleza, cada tejido puede recibir cantidades diferentes del fármaco, el cual, además, pasará allí tiempos variables.
Metabolismo o biotransformación
La transformación puede consistir en la degradación (oxidación, reducción ohidrólisis), donde el fármaco pierde parte de su estructura, o en la síntesisde nuevas sustancias con el fármaco como parte de la nueva molécula (conjugación). La oxidación se realiza fundamentalmente por lasisoenzimas del citocromo P450 en lo que se conoce como metabolismo de fase I. La conjugación es la fase fundamental delmetabolismo de fase II, pudiendo existir una tercera fase o metabolismo de fase III, característica de los organismos pluricelulares.
En el humano y en la mayoría de los mamíferos, el metabolismo de los fármacos se realiza fundamentalmente a nivel del hígado. Como resultado de la biotransformación se obtienen nuevas sustancias que reciben el nombre demetabolitos. Los metabolitos pueden mantener la capacidad del fármaco original para ejercer sus efectos, o bien haberla vista disminuida, aumentada o incluso haber cambiado sus efectos por otros distintos. Por ello se habla de metabolitos activos, o inactivos. Incluso, en ocasiones el fármaco no presenta actividad farmacológica alguna, siendo alguno de sus metabolitos los que realmente ejercen su actividad. Se habla en este caso de profármacos, y un ejemplo claro son algunas estatinas(simvastatina y lovastatina). Evidentemente, los profármacos dependen del buen funcionamiento del metabolismo para poder ejercer de forma adecuada sus efectos.
En ocasiones los propios fármacos o algunos de sus metabolitos son capaces de modificar la capacidad metabólica de las enzimas, aumentando o disminuyendo su actividad.
![]()
![]()
![]()
![]()
![]()